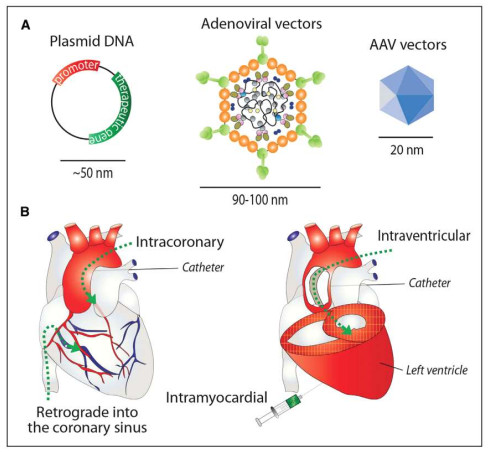
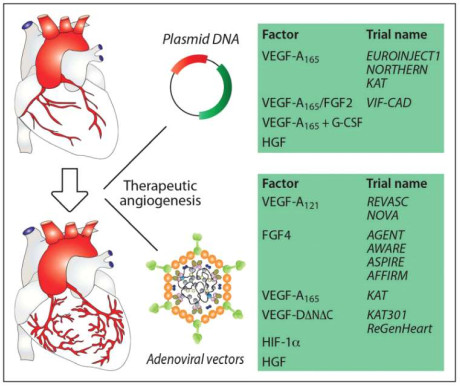
SCENARIO 2: CGTS FOR PREVALENT HEART DISEASE INDICATIONS
For more prevalent heart disease indications, the value proposition, price, and adoption of CGTs change intensely. They fall into three broad categories:
Uncertainty in the regulatory environment and high expectations for product performance
The majority of drug development for cardiovascular diseases has typically been accomplished through large-scale outcomes studies, where a survival advantage must be demonstrated compared to the standard of care and safety risks are essentially not tolerated. Endpoints that focus on functional improvements, such as ejection fraction (EF) and 6-minute walk tests (6MWT), have not been accepted as sufficient for approval. This necessitates the use of very long and expensive randomized and placebo-controlled clinical studies. For example, one report suggests that the average size of a clinical study used to back recommendations for heart failure treatments included more than 2,300 patients, with one study having as many as 8,400 participants.
Investigations for remedies meant to tackle diabetes may necessitate very stringent assessments involving 5,000-15,000 patients to exclude cardiovascular danger. This could at least partially explain why the development of drugs for cardiovascular ailments has been hard. Many studies have determined that the total likelihood of successful drug production from Phase I through commercial launch is 4-7%, one of the lowest in all therapeutic areas. Recent analysis has indicated that, usually, biopharmaceutical companies use around $1B in clinical progress for every cardiovascular product authorization, the greatest proportion in comparison to any other therapeutic area. Some specialists think that clinical tests in cardiovascular medicine have turned out to be larger, more expansive and intricate, resulting in some pharmaceutical companies diverting away from the cardiovascular sector.
Low-cost benchmarks for standard of care treatments
First line therapies for heart failure have mostly been the realm of generic small molecules, including angiotensinconverting enzyme inhibitors (ACEI), angiotensin II receptor blockers (ARBs), beta blockers (BBs), aldosterone antagonists (AldA), and diuretics. These medications are very well accepted as safe and effectve, with proven longterm survival benefts. They are also very inexpensive. One of the most widely prescribed ACEi drugs has an annual cost of less than $500 per year, and this combination of generic first line therapies has a collective annual cost less than $2,000 per year. These therapies are considered very cost effective, and in some scenarios these medications save costs (i.e. where heart failure patents’ lives were prolonged at lower costs to the healthcare system).
Reasonable standards for conventional healthcare
Generic small molecules, such as angiotensin-converting enzyme inhibitors (ACEI), angiotensin II receptor blockers (ARBs), beta-blockers (BBs), aldosterone antagonists (AldA), and diuretics, have typically been the first-line treatments for heart failure. These drugs are widely recognized as effective and safe, with long-term survival benefits that have been demonstrated. They are also reasonably priced. These treatments are seen as being very cost-effective, and in certain cases they result in cost savings.
High price sensitivity
Heart failure is a leading cause of death and a major expense for payers, both private and public. It is estimated that the total direct and indirect costs of heart failure will reach $70 billion by 2030. With the rise in costs, payers are increasingly focusing on cost control for this therapeutic area. Most cases of heart failure are observed in individuals aged 65 or older due to aging-related weakening of the heart muscle, as well as chronic diseases that can lead to heart failure. As such, most heart failure patients are covered by medical care organizations, the primary public option. Hospital stays are a common occurrence for these patients, making heart failure one of the most expensive chronic diseases to treat.
Our gene therapy technology consists of three core elements:
- A "gene cassette" containing a therapeutic gene is used to rectify a malfunctioning or absent protein, thus restoring normality or curtailing a disease-associated effect in targeted cells.
Therapeutic Gene Cassettes
Constructing our own gene therapies, we introduce a transgene into cells for the production of a beneficial protein or RNA structure. This transgene is contained in a gene cassette with DNA promoters that coordinate its expression in particular tissues.
- A vector delivery system based on adeno-associated viruses (AAVs) to transport the genetic cassette.
Best-in-Class AAV-based Vector Delivery System
Our product candidates, developed for hemophilia B and Huntington's disease, rely on the AAV5 variant, or serotype, of the vector delivery system. This particular type of adeno-associated virus has been utilized in pre-clinical research and more than 80 clinical trials, where it has displayed an excellent safety profile. Furthermore, studies have demonstrated that a single treatment with AAVs can lead to long-term therapeutic gene expression. We hold exclusive, worldwide rights to AAV5 for use in therapeutic products delivered to the brain or liver, and our investigations suggest that this vector can provide greater efficacy in treating patients when compared to other AAV-based gene therapies.
Clinical trials involving nearly 80 patients with hemophilia B and other conditions have demonstrated that gene therapies based on AAV5 are safe and well-tolerated. No individual receiving the treatment in these trials has exhibited a confirmed T-cell-mediated immune response towards the capsid. Furthermore, pre-clinical and clinical data suggest that AAV5-based gene therapies may be an effective treatment for patients with existing antibodies to AAV5, potentially increasing the number of people eligible for this type of therapy.
The presence of circulating anti-AAV neutralizing antibodies, which can be present in patients beforehand and may inhibit successful gene transfer, is one of the main difficulties in AAV-based gene therapy. Preclinical evidence demonstrating successful and efficient AAV5 transduction in non-human primates with pre-existing anti-AAV5 neutralizing antibodies (NABs) has been provided. Existing anti-AAV5 antibodies did not impair the efficiency of the AAV5 vector's transduction at any of the measured levels. It indicates that patients with pre-existing anti-AAV5 NABs may still be able to be successfully treated with AAV5 gene therapies, which offers a far wider potential group of eligible patients for AAV5-based gene treatments than was previously anticipated.
We have made significant headway in improving and advancing re-administration and cross-administration strategies which we think will have a great effect on the usage of our gene therapies and possibly permit multiple administrations. We have presented preclinical results indicating successful re-administration of gene therapy with our AAV5 vector following a specific immunoadsorption technique in non-human primates (NHPs). In addition, we have published information demonstrating profitable cross-administration of gene therapies in NHPs using sequential administration of AAV5 and AAV1 vector serotypes, implying that cross-administration of AAV5 gene therapies with other vectors might be feasible in humans. Despite this progress, high levels of circulating anti-AAV neutralizing antibodies can develop after a single administration of gene therapy and can prevent successful gene transfer in patients. AAV5 is a premier vector with the capacity to deliver gene therapies more efficiently and securely to a larger number of patients in need of care.
- Implementing administrative strategies to efficiently transport the appropriate gene into the liver or central nervous system.
Administration Technology
We and our partners are making great strides in harnessing a range of technologies to deliver our gene therapies to the right tissues and organs for the desired outcome. This includes methods such as intravenous infusion for our hemophilia B program or MRI-guided injection into the heart for our heart disease program. With our competent team and cutting-edge facilities, we have all the necessary components to create a successful gene therapy product.
The platform is intended to be modular, which may enable us to quickly develop, produce, and obtain regulatory approval for a variety of gene therapies using essentially the same basic building blocks. We anticipate that in certain circumstances, the only element that has to be altered to target a new disease in a particular tissue is the disease-specific gene cassette. As a result, we might be able to considerably reduce overall development risk, time, and expense while also reducing the amount of preclinical and possibly clinical testing needed to achieve regulatory approval.
Improvement in Gene Therapy Technology
Our platform for next-generation gene silencing is our own, exclusive technology. It is intended to inhibit the entire target organ by secondary exosome-mediated transport and degrade disease-causing genes without producing off-target damage. We have developed novel therapeutic structures for gene therapy candidates that can be administered by AAVs and may have long-lasting action. Preclinical research on gene treatments based on this technology has revealed several significant benefits, such as better tissue-specificity, improved nuclear and cytoplasmic gene suppression, and no off-target consequences linked to impact on the cellular transcriptome.